Development of Tools
Development of Transsynaptic Tracers
Eugene Drokhlyansky, Graduate Student
Didem Goz, Postdoctoral Fellow
James DeMelo, Research Technician
Xiang Ma, Postdoctoral Fellow
Jonathan Tang, Graduate Student
Nathan Mundell, Postdoctoral Fellow
One goal of neuroscientists is to discover and understand the circuits that define the various functions carried out by the nervous system. To this end, we developed a new viral tracer, based upon the vesicular stomatitis virus (VSV) (Beier et al. 2011, 2013a). We demonstrated that VSV can travel between synaptically connected cells in many types of organisms, including mammals, birds, fish, and amphibians (Submitted). We were also able to direct its transmission either anterograde or retrograde using different types of viral glycoproteins. Furthermore, we could alter the properties of the VSV so that it crossed one synapse and then stopped transmitting, or continued to transmit across several synapses. We used these vectors to begin to examine the direction-selective circuits in the mouse retina (Beier et al. 2013b). Previously unknown synaptic partners were detected, and their connections were confirmed using electrophysiology, in collaborative studies (Figure 1).

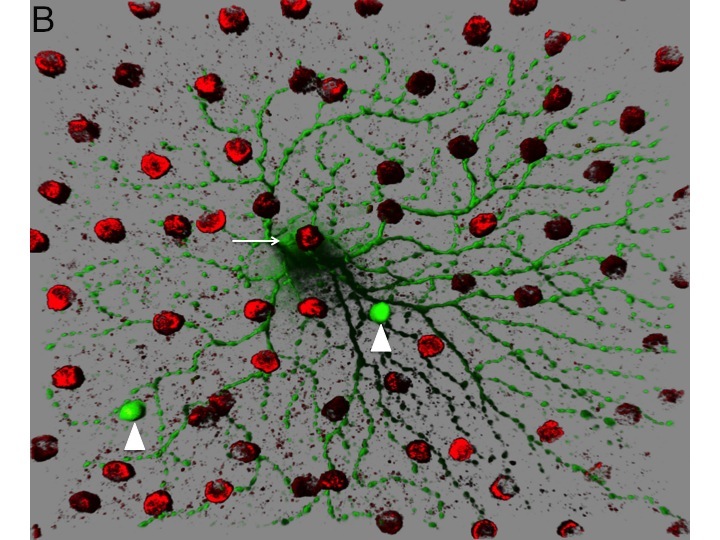
Figure 1. Circuitry tracing using VSV. A. VSV can be engineered to discover the types of cells that provide presynaptic input to initially infected cells. VSV virions with two types of glycoproteins on the surface can be created in tissue culture. One type of glycoprotein, the rabies virus glycoprotein (RABV-G), is supplied by the tissue culture cells. Another type, a fusion of the extracellular domain of the avian EnvA glycoprotein and the cytoplasmic domain of RABV-G, is encoded by the VSV genome. The virus preparation is injected into the lateral geniculate nucleus (LGN), a retinorecipient area. Virions can enter the terminals of retinal ganglion cells (RGCs) and travel to the cell body, where they can replicate, and produce GFP. The viral particles produced by the infected RGCs will have on their surface only the EnvA fusion protein, due to the fact that only this glycoprotein is encoded within the VSV genome. The viral particles will exit from the soma and dendrites and infect only presynaptic cells which express TVA, the receptor for EnvA. The Chat-Cre mouse was crossed to a floxed TVA strain (Beier et al. Dev. Biol. 353: 309-320, 2011), as well as a floxed tdtomato strain (Ai9 created by the Allen Brain Institute). The Chat gene is only expressed in cholinergic cells, which in the retina, are the starburst amacrine cells (SACs). By tracking which types of GFP+ RGCs transmit to the SACs, one can infer the type of RGCs that are postsynaptic to SACs. B. An image of an area of a flat mount of a retina infected as described in A. The GFP+ cell is an OFF-alpha retinal ganglion cell and the red cells are SACs labeled by tdtomato. Two SACs were infected by VSV transmitted by the OFF-alpha ganglion cell.
GFP as a Scaffold for Biological Activities
Many transgenic organisms express GFP in specific cell types. To expand the utility of these lines to include biological perturbations only in GFP-positive cells, we developed a method to use GFP as a scaffold (Tang et al. 2013). This method relies on GFP-binding proteins (GBPs), derived from Camelid antibodies previously characterized by Ulrich Rothbauer and colleagues. We used pairs of GBPs as fusion partners for protein domains that, in the presence of GFP, would constitute a biological activity. For example, one GBP fusion that binds to a specific DNA sequence can be used in combination with another that activates transcription. GFP can bring these two fusions together to activate transcription from a specific promoter (Figure 2, 3). This version of the method is referred to as T-DDOG, for transcription devices dependent on GFP. Another set of fusions reconstitutes Cre recombinase, allowing the manipulation of gene structure only in GFP-positive cells, and is referred to as Cre-DOG, for Cre dependent upon GFP (Tang et al. 2015) (Figure 4). These constructs are modular and are relatively straightforward to engineer to create many types of activities. We are currently using GFP to create other types of GFP-dependent activities.
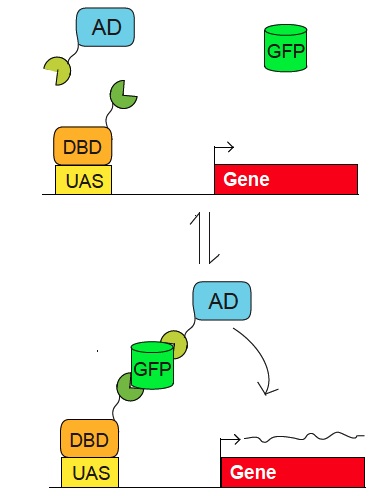
Figure 2. Scheme for use of GFP as a scaffold to control biological activities. Pairs of fusion proteins that bind to GFP (in green and orange) can activate a reporter whose regulatory region (e.g. UAS) is recognized by a specific DNA binding domain (DBD). Reporter activation (up to 200 fold) is dependent upon GFP. The reporter plasmid can encode virtually any gene, such as Cre, channelrhodopsin, etc. The system also works with GFP derivatives, such as YFP. The modularity of the components allows for the use of different DBDs, and has so far been developed for the tet, lexA, and Gal4 DBDs and several different activation domains (AD). (Tang et al. 2013).
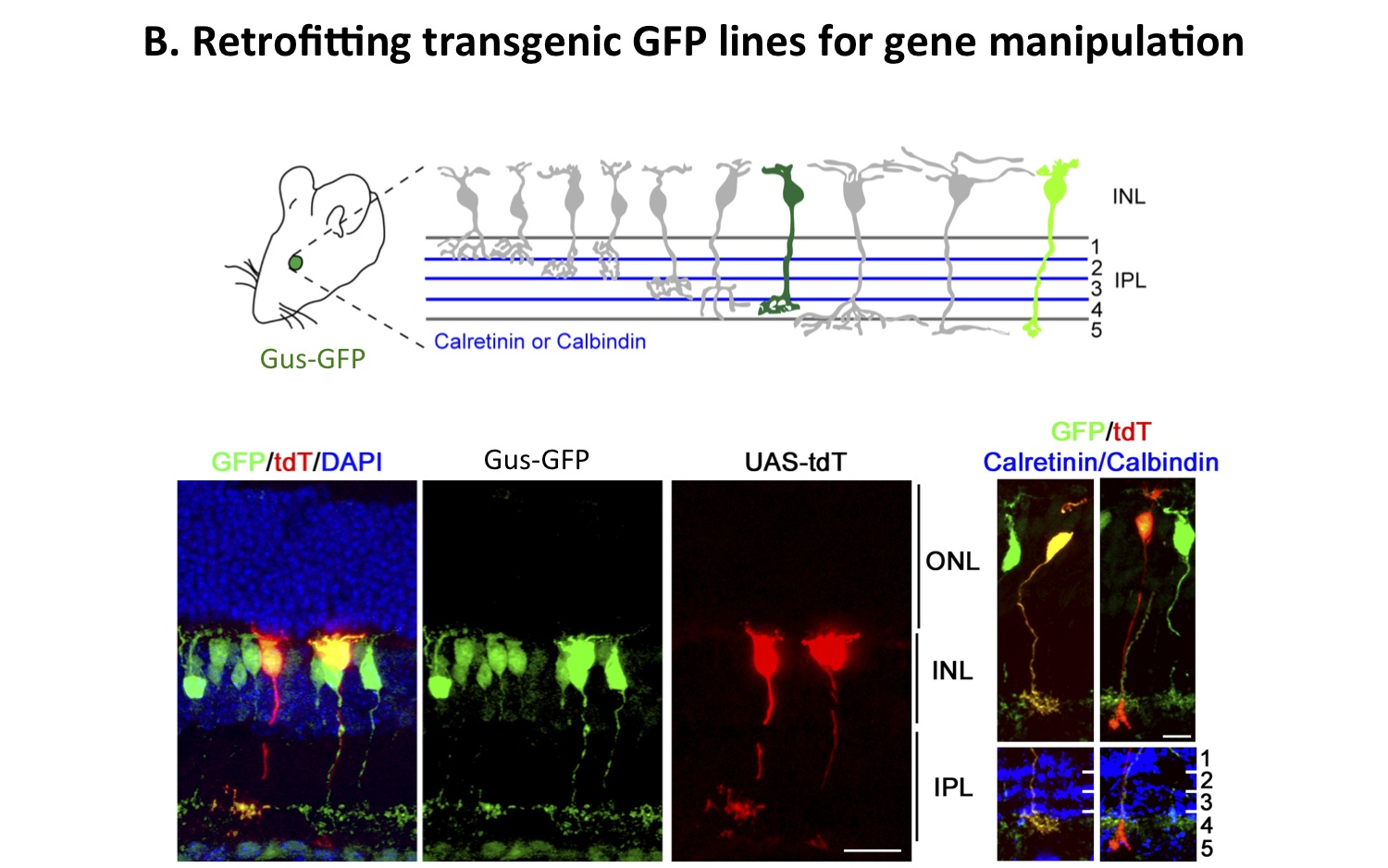
Figure 3. Retrofitting a transgenic GFP mouse line for GFP-dependent manipulation of gene expression and neural circuit activity. The use of GFP as a scaffold to create biological activities only in GFP-expressing cells in a transgenic mouse is shown. Tg(GUS8.4GFP) expresses GFP in type 7 retinal cone bipolar and rod bipolar cell types (green) of the retina. A schematic of the types of bipolar cells in the retina is shown. Electroporation of GFP-binding proteins fused to transcription activation and DNA binding domains can lead to the expression of specific genes, here a red fluorescent protein, in a subset of bipolar neurons in the murine retina. Two tdT-expressing bipolar cells are shown, with the Type 7 shown on the left and a rod bipolar on the right, labelled by the transcription of a UAS-tdT construct driven by the GFP-dependent fusion proteins (as shown in A). Axonal stratification determined by anti-Calretinin (left) or anti-Calbindin (right) staining shows that the cellular morphology and identity were not perturbed. (Tang et al. 2013).
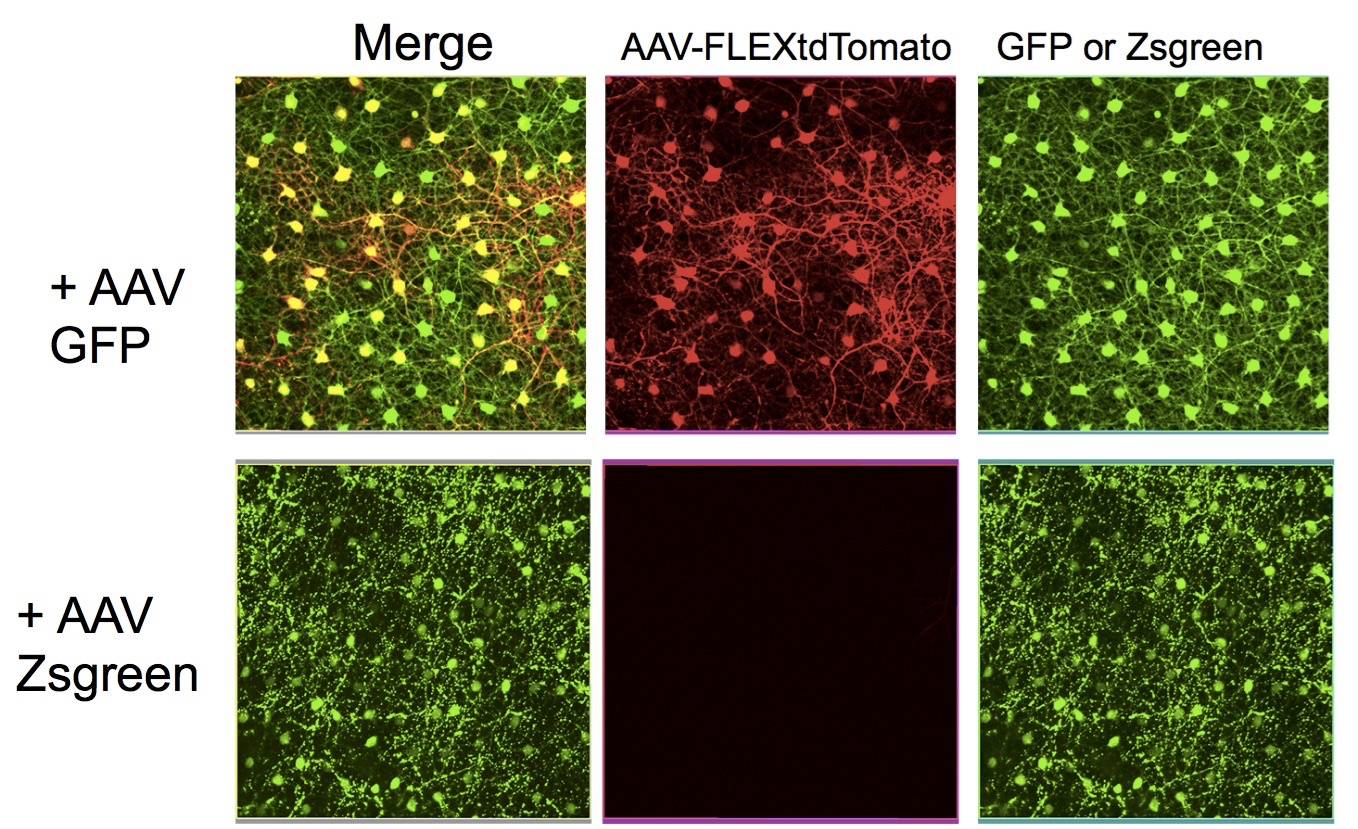
Figure 4. Cre-DOG activity can be delivered in vivo using AAV vectors. An active Cre recombinase was reconstituted using two nanobody fusion proteins, as in Figure 3. One anti-GFP nanobody was fused to the N-terminal portion of Cre, while another anti-GFP nanobody was fused to the C-terminal portion. These two fusion genes were encoded by AAV vectors, which were delivered to the murine retina in vivo. Along with the AAV vectors encoding the nanobody fusion proteins, an AAV encoding GFP, or an AAV encoding Zsgreen, were delivered. To visualize Cre activity, an AAV encoding a Cre-dependent tdtomato also was injected. The retina was then imaged for GFP and tdtomato at the level of the horizontal cells. The presence of GFP led to the activation of Cre, which led to tdtomato expression, while the presence of Zsgreen, which is not recognized by the anti-GFP nanobodies, did not lead to Cre activation.
Electroporation
Claudio Punzo (former Postdoctoral Fellow (now at UMassachusetts Medical School)
Electroporation can be carried out in vivo or in vitro, on chick, mouse, or rat retinas. This method has advantages over that of viral transduction and over trangenic animal production. Several (at least 5) plasmids can be simultaneously delivered, with regulated expression from each of the plasmids. A solution with a high concentration of plasmids is injected into the subretinal space and short pulses of high voltage are applied. The DNA is likely passively taken up via transient holes created in the membrane by the high voltage. Thus, only cells immediately adjacent to the injection site are transduced, and there appears to be no specificity in the uptake. For postnatal day 0-7 mouse and rat retinas, this means that progenitors that make rods, bipolars, amacrines and Muller glial cells are transduced, along with newly postmitotic cells that will achieve these fates (Figure 5). We have also been able to deliver plasmids in vivo to embryonic mouse retinas, with the most bright labeling of cells that become postmitotic shortly after deliver (e.g. cones, horizontal cells, amacrine cells). We have been unsuccessful transducing cells from the vitreal side, or in transducing adult cells from the scleral side. Electroporation of neonatal mouse and rat pups leads to expression for at least 50 days. Electroporation of rapidly dividing early chick retinal cells, e.g. at stage 10, leads to expression for only 2-3 days, likely due to dilution, and possibly degradation, of the plasmids. Plasmids encoding genes for gain-of-function studies have been successfully used. In addition, plasmids encoding RNA hairpins for RNAi have been successfully used. For studies of cis-acting regulatory regions, we have also been able to use plasmids with regulated promoters to begin to determine the sequences required for proper cell type specific expression (Figure 6).
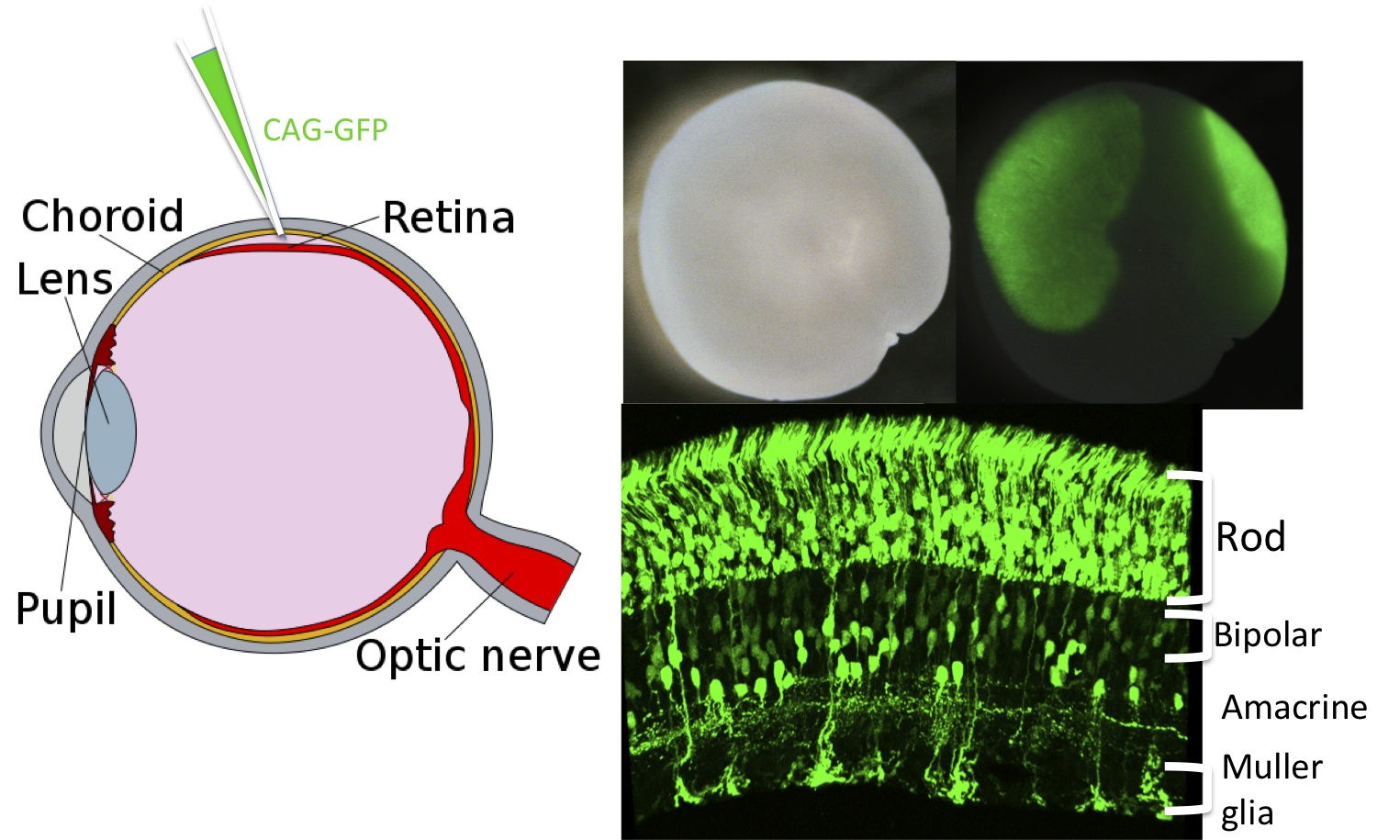
Figure 5. Electroporation of the retina with multiple plasmids.
Plasmids encoding a variety of proteins, shRNAs, or enhancer constructs can be introduced into the retina via electroporation in vivo. Here a plasmid encoding GFP, driven by CAG, a broadly active promoter, was introduced by subretinal injection at postnatal day 0. Several weeks later, the retina was removed and imaged to show where the GFP fluorescence was induced. Sections through the retina showed expression in rods, bipolar and amacrine interneurons and Mueller glia.
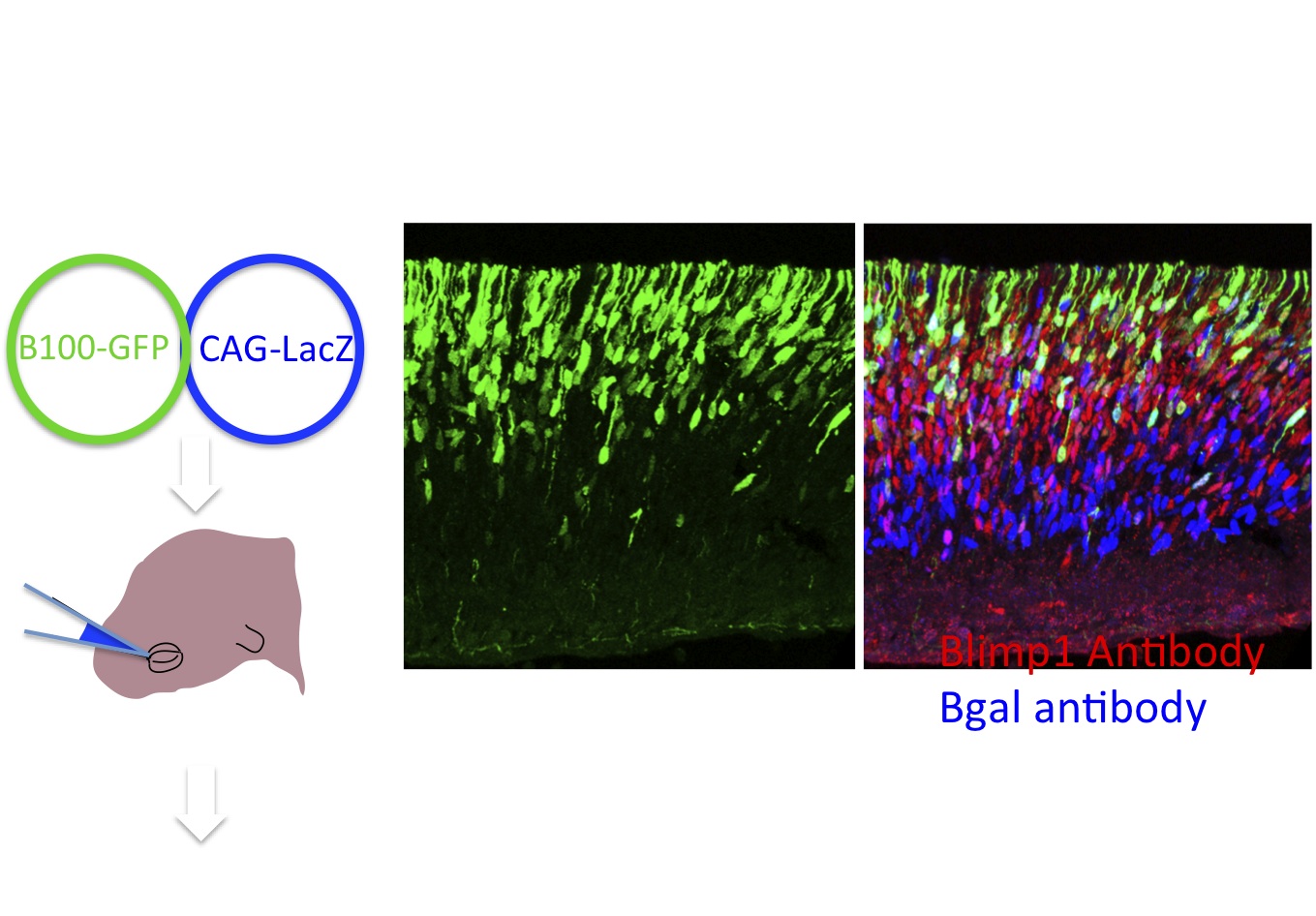
Figure 6. Electroporation is a rapid way to assess regulatory regions. Potential cis-regulatory regions are cloned into a plasmid, Stagia 3 (Billings et al. 2012), that expresses GFP or human placental alkaline phosphatase from a simple TATA element, only if the enhancer has activity in the retina. Electroporation into the retina in vivo or in explants allows for a rapid assay of enhancer activity. Here an example of a regulatory element, B108, of only 108 base pairs from the Blimp1 gene is shown. This enhancer recapitulates endogenous Blimp1, as shown by the colocalization of GFP and anti-blimp1 immunohistochemistry. Co-electroporation of a plasmid encoding lacz driven by CAG provides a marker for cells that take up plasmid, as evidenced by anti-bgal immunohistochemistry. (Data and images from Sui Wang, Wang et al. Dev. Cell. 2014)
Plasmids created by T. Matsuda for use in electroporation studies are available from Addgene (http://www.addgene.org/).
Lineage analysis using retroviral vectors
Knowledge of the geneological relationships of cells during development can allow one to gain insight into when and where developmental decisions are being made. Geneological relationships can be revealed by a variety of methods, all of which involve marking a progenitor cell and/or a group of cells and then following the progeny. We have used retroviral vectors to mark individual progenitor cells with genes that can be detected histochemically (Figure 7) . Gamma retroviruses can only integrate, and thus express, in mitotic cells, and they can be delivered at low concentration to allow for the identification of clones. These features provide for unambiguous marking of the progeny of individual progenitor cells, which are features that are typically not allowed by e.g. cre fate mapping. We augmented these original vectors with bar codes that can be sequenced from infected cells to allow assignment of sibling relationships among scattered cells (Figure 8) (Walsh and Cepko, 1992; Golden et al. 1995). We have also generated bardcoded libraries of viruses that express recombinases (Rabe and Cepko, unpublished). Viruses can be directed to cells that express the avian receptor, TVA, to allow the assignment of fates in a clonal manner to cells expressing TVA, as arranged by crossing a conditional TVA allele to a Cre of choice (Beier et al. 2011). Finally, we have made viruses that report recombinase activity through the use of floxed alleles within the virus (Surzenko and Cepko, unpublished).

Figure 7. Clones in the rat and mouse retina marked by infection with retrovirus vectors. A, B. Clones marked by infection in vivo in the rat retina on the day of birth. Retinal progenitor cells so marked gave rise to clones of multiple cell types, indicating that they were multipotent. Clones include both neurons and glia C. A clone from infection of the embryonic mouse retina, showing larger clones and more complexity than clones initiated in the postnatal period. From Turner and Cepko, 1987; Turner et al. 1990.
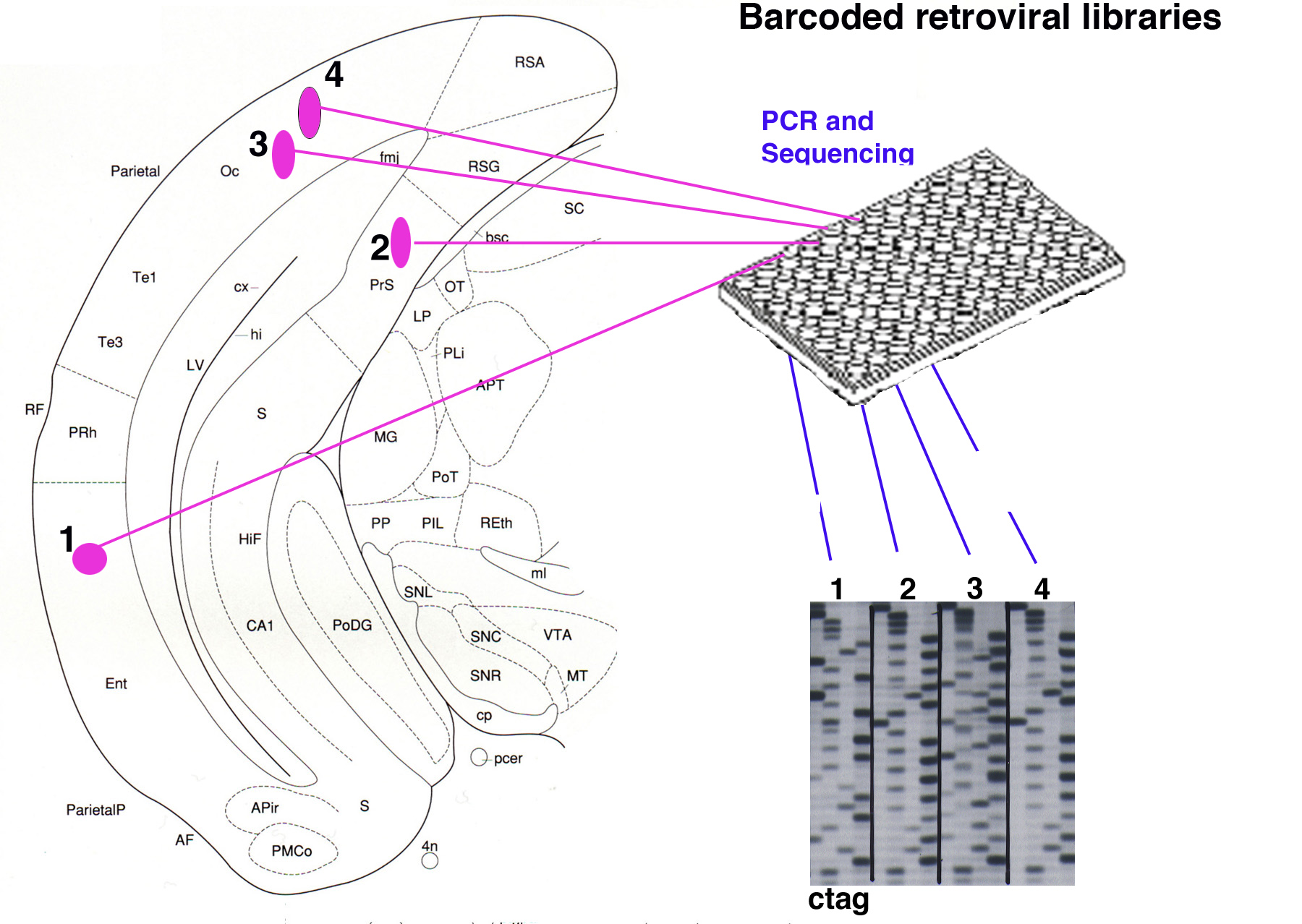
Figure 8. Retroviral libraries with bar codes allow unambiguous clonal assignments. A retroviral library encoding a histochemical or fluorescent reporter gene can also be made to encode >10^5 distinct oligo’s, or bar codes. After infection into the brain, sections are processed for histochemical detection of virally infected cells. Each cell is then excised and processed for PCR for the bar code, which is then sequenced. Cells that share the same viral bar code are highly likely to be sibling cells due to the high library complexity.